- Снижение уровня Hb < 135 г/л для мужчини, <120 г/л для женщин
- Снижение уровня гематокрита < 40% у мужчини, < 36% у женщин
- Снижение количество эритроцитов ниже 4,0 млн. в 1 мм3 для мужчин, ниже 3,7 млн. в 1 мм3 для женщин
- Увеличение среднего объёма эритроцитов (MCV) > 100мкм3
- Увеличение среднего содержания Hb в эритроцитах (MCH) > 35пг
- Увеличение цветового показателя >1,1
- Увеличение количества макроцитов (больших, овальных эритроцитов диаметром >100 мкм в периферической крови, появление мегалоцитов–эритроцитов диаметром более 120 мкм. Сдвиг кривой Прайс–Джонса вправо
В костном мозге: мегалобластический тип кроветворения
1. «Раздражённый» красный росток: соотношение миелоидного и эритроидного ростков 1:3 при норме 3:1
2. Появление мегалобластов и гигантских метамиелоцитов
В периферической крови:
1. Изменение морфологии эритроцитов (пойкилоцитоз, ядерные формы, остатки ядра – тельца Жоллии, кольца Кебота)
2. Гиперсегментация нейтрофилов
3. Лейкопения, тромбоцитопения, ретикулоцитопения
4. Наличие мегалобластов (не всегда)
Вопрос 21. Анемии при недостатке фолиевой кислоты. Этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
| Этиология фолиеводефицитной анемии
|
| Недостаточное потупление фолиевой кислоты с пищей:
Голодание;
Вскармливание грудных детей козьим молоком;
Отсутствие зелёных овощей в пищевом рационе;
Длительная термическая обработка пищи.
Нарушение всасывания и утилизация витамина и утилизация фолиевой кислоты:
У недоношенных детей;
дефицит витамина В12;
патология тонкого кишечника (энтерит, полипоз, тропическая спру, глютеновая энтеропатия, резекция, рак и др.);
алкоголизм;
приём лекарственных препаратов (оральных контрацептивов, противосудорожных, противотуберкулёзных препаратов и др.).
Повышенное расходование фолиевой кислоты:
В физиологических условиях (при беременности, лактации, в пубертатном периоде);
В условиях патологии — при заболеваниях с высокой скоростью процессов пролиферации клеток (гемолитические анемии, множественная миелома, сублейкемический миелоз), туберкулёзе и др.
Нарушение депонирования фолиевой кислоты:
(при токсическом и вирусных гепатитах, циррозе печени, гепатоцеллюлярном раке и др.)
|
Клиника схожая с таковой анемией Аддисона-Бирмера
Вопрос 22. Ахрестические анемии. Этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
B12 (ФОЛИЕВО) - АХРЕСТИЧЕСКАЯ АНЕМИЯ
Под названием «ахрестическая анемия» впервые Israels и Wilkinson (1936) описана своеобразная гиперхромная мегалоцитарная анемия, отличающаяся от классической пернициозной анемии по своему патогенезу и рефрактерности к печеночной терапии.
Эта анемия была названа ахрестической, т. е. «анемией от неиспользования». Этим подчеркивалось, что анемия возникает вследствие того, что костный мозг не в состоянии использовать имеющиеся в организме антианемические субстанции (витамин B12, фолиевую кислоту). Печень умерших от ахрестической анемии содержит витамин В12 и фолиевую кислоту, но эти витамины не усваиваются костным мозгом. В настоящее время это состояние рассматривается как предлейкоз.
Клиника. В отличие от болезни Аддисона—Бирмера при В12(фолиево)-ахрестической анемии отсутствуют признаки поражения пищеварительной и нервной системы: нет глоссита, нет ахилии, желудочный сок содержит соляную кислоту и пепсин; не наблюдается диспепсических явлений, в частности поноса. Не выражены и симптомы повышенного распада крови: нет желтушности, печень и селезенка не увеличены.
Картина крови. Кровь соответствует картине пернициозной анемии в период рецидива; количество эритроцитов падает до 1 000 000 и ниже, цветной показатель выше единицы. Среди эритроцитов преобладают макро- и мегалоциты; последние составляют до 20—30% всех эритроцитов. Ретикулоцитоз низкий. Осмотическая резистентность эритроцитов нормальная. Количество лейкоцитов нормально или уменьшено (за счет гранулоцитопении). В костномозговом пунктате обнаруживается картина мегалобластического кроветворения (мегалобласты в различных стадиях созревания). Нормобластический эритропоэз угнетен.
Патогенез. Отсутствие симптомов повышенного разрушения эритроцитов исключает роль гемолиза в патогенезе ахрестической анемии.
При ахрестической анемии нет поражения кишечника, следовательно, всасывание антианемических веществ не нарушено, нет и нарушений желудочной секреции — желудочный (внутренний) антианемический фактор сохранен (что доказывается положительной крысоретикулоцитарной реакцией).
Содержание витамина B12 в плазме крови нормально или повышено.
Доказано, что печень умерших от ахрестической анемии обладает антианемической активностью, следовательно, в ней содержатся витамин B12 и фолиевая кислота.
Эти факты дают основание высказать предположение, что при B12-(фолиево)-ахрестической анемии витамины кроветворения всасываются в кишечнике и откладываются в печени, но не утилизируются костным мозгом.
Течение болезни неуклонно прогрессирующее, без ремиссий. Болезнь длится около 1,5—2 лет.
Прогноз неблагоприятный.
Вопрос 23. Железодефицитная анемия. Этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
ЖДА составляют 70% всех анемий. По патогенетическому принципу с учетом основных этиологических причин железодефицитные анемии делят на пять основных подгрупп (Л.И. Идельсон):
1) связанные с повышенной потерей железа;
2) связанные с недостаточным исходным уровнем железа;
3) связанные с повышенным расходованием железа;
4) связанные с нарушением всасывания железа и недостаточным поступлением его с пищей (алиментарные);
5) связанные с нарушением транспорта железа.
Причины и механизмы развития железодефицитных состояний
| Этиологические факторы
| Характеристика
| Патогенез
|
| Особенные периоды жизни
| Дети недоношенные и новорожденные
Дети первых лет жизни
| Недостаточный исходный уровень железа
|
| Интенсивный рост (пубертатный период)
Беременность
Лактация
| Повышенное расходование железа
|
| Патологические состояния
| Хроническая кровопотеря: при частых лечебных кровопусканиях, донорстве;
При заболеваниях ССС (гипертоническая болезнь, геморрагическая телеангиэктазия и др.);
При патологии ЖКТ (варикозное расширение вен пищевода, диафрагмальная грыжа, язва желудка и 12-перстной кишки, язвенный колит, дивертикулёз, геморрой и др.);
Из органов мочеполовой системы (алкогольная нефропатия, туберкулёз почек, почечнокаменная болезнь, полипы и рак мочевого пузыря, обильные меноррагии, эндометриоз, миома матки и др.);
Из органов дыхательной системы (рак лёгкого, туберкулёз, бронхоэктазия и др.);
При заболеваниях системы крови (лейкозы, апластическая анемия и др.);
Припатологии системы гемостаза (аутоиммунная тромбоцитопения, гемофилии, ДВС-синдром и др.)
| Повышенная потеря железа
|
| Патологические состояния и болезни
| Патология ЖКТ:
Резекция желудка и кишечника;
Гипосекреция желудочного сока;
Хронический энтерит;
Дисбактериозы;
Глистные инвазии и др.
| Нарушение всасывания железа
|
| Наследственная атрансферринемия
Приобретённая гипотрансферринемия (при нарушении белоксинтезирующей функции печени)
| Нарушение транспорта железа
|
| Алкоголизм
| Комбинация факторов:
Недостаточное поступление железа; нарушение транспорта железа; нарушение всасывания железа; потеря железа
|
| Нарушение всасывания железа
| Нерациональное питание:
Голодание;
Вегетарианская диета;
Искусственное вскармливание грудных детей
| Недостаточное поступление железа
|
| Избыточные физические нагрузки
| Повышенное расходование железа
|
Клиническая картина
1.Анемический синдром.
2.Сидеропенический синдром характеризуются гипоксическим синдромом, проявления которого зависят от степени снижения гемоглобина. Характерны изменения кожи и слизистых оболочек: сухость и зуд кожи, выпадение волос, ломкость ногтей и изменение их формы (койлонихии - ложкообразные ногти); из-за атрофии сосочков языка развивается глоссит и изменяется вкусовая чувствительность (дети с удовольствием вместо конфет грызут мел, уголь, глину или лед), часто возникает ангулярный стоматит («заеды»), могут быть диспепсические расстройства и признаки эзофагита. У больных ЖДА снижается иммунитет и присоединяются инфекционные заболевания.
Клинический анализ крови
В общем анализе крови при ЖДА будут регистрироваться снижение уровня гемоглобина и эритроцитов. Умеренная эритроцитопения может проявляться при Hb <98 г/л, однако снижение эритроцитов <2·1012/л для ЖДА не характерно. При ЖДА будут регистрироваться изменения морфологических характеристик эритроцитов и эритроцитарных индексов, отражающих количественно морфологические характеристики эритроцитов.
Морфологические характеристики эритроцитов. Размер эритроцитов — нормальный, увеличенный (макроцитоз) или уменьшенный (микроцитоз). Для ЖДА характерно наличие микроцитоза. Анизоцитоз — различия в размерах эритроцитов у одного и того же человека. Для ЖДА характерен выраженный анизоцитоз. Пойкилоцитоз — наличие в крови одного и того же человека эритроцитов разной формы. При ЖДА может быть выраженный пойкилоцитоз. Цветовой показатель эритроцитарных клеток (ЦП) зависит от содержания в них гемоглобина. Возможны следующие варианты окрашивания эритроцитов:
· нормохромные эритроциты (ЦП= 0,85-1,05) — нормальное содержание гемоглобина в эритроцитах. Эритроциты в мазке крови имеют равномерную розовую окраску умеренной интенсивности с небольшим просветлением в центре;
· гипохромные эритроциты (ЦП<0,85) — содержание гемоглобина в эритроците снижено. В мазке крови такие эритроциты имеют бледно-розовую окраску с резким просветлением в центре. Для ЖДА гипохромия эритроцитов является характерной и часто сочетается с микроцитозом;
· гиперхромные эритроциты (ЦП>1,05) — содержание гемоглобина в эритроцитах повышено. В мазке крови эти эритроциты имеют более интенсивную окраску, просвет в центре значительно уменьшен либо отсутствует. Гиперхромия связана с увеличением толщины эритроцитов и часто сочетается с макроцитозом;
· полихроматофилы — эритроциты, окрашенные в мазке крови в светло-фиолетовый, сиреневый цвет. При специальной суправитальной окраске это — ретикулоциты. В норме могут быть единичными в мазке.
Анизохромия эритроцитов — различная окраска отдельных эритроцитов в мазке крови.
Вопрос 24. Апластические анемии. Этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
Апластические анемии — гетерогенная группа заболеваний системы крови, основу которых составляет уменьшение продукции клеток костного мозга, чаще трех клеточных линий (эритроцито-, лейко- и тромбоцитопоэза).
Причинами апластической анемии могут быть:
· Химические вещества (мышьяк, соли тяжёлых металлов).
· Ионизирующее излучение. (см. Мария Склодовская-Кюри)
· Лекарственные препараты (НПВС, цитостатики, мерказолил, анальгин).
· Инфекционные агенты (вирусы, м/о).
· Аутоимунные процессы (СКВ, синдром Ширена).
Патогенез
Механизм остановки пролиферации полипотентных стволовых клеток остается неясным. Предполагается первичное нарушение функции стромального микроокружения, обеспечивающего стволовые клетки ростовыми факторами. Однако более вероятной представляется активация факторов внеклеточной иммунной супрессии, направленных на полипотентные стволовые клетки. В пользу последнего механизма говорят благоприятные результаты, полученные при применении антииммуносупрессивных препаратов у значительного числа больных. На возможность генетической предрасположенности к развитию апластической анемии указывает высокая частота выявляемых при АА антигенов 11 класса системы DR-2 и DPw3. Известен ряд этиологических факторов: некоторые лекарственные средства, химические соединения, ионизирующая радиация и вирусные инфекции, способных вызвать заболевание. Эти апластические анемии рассматриваются как вторичные и обладают более благоприятным прогнозом. При отсутствии достоверных данных об этиологии заболевания апластические анемии обозначаются как идиопатические, имеют более неблагоприятный прогноз и встречаются у 65% больных.
Классификация апластических анемий.
I. Идиопатические апластические анемии.
1. врожденная (анемия Фанкони)
2. приобретенная
II. Вторичные апластические анемии
a. вследствие воздействия лекарственных и химических веществ:
b. лекарственные средства (хлорамфеникол, сульфаниламиды, пирозолоны, препараты золота и другие)
c. химические вещества (бензол и его производные, инсектициды и другие)
d. инфекционных и вирусных агентов (вирусный гепатит, милиарный туберкулез, сепсис и другие)
e. метаболические (панкреатиты, беременность)
III. иммунологические (аутоиммунные, при реакции трансплантат против хозяина)
IV. при пароксизмальной ночной гемоглобинурии
КЛИНИКА
Больные апластическими анемиями бледны, с сохраненным или избыточным подкожным жировым слоем. Они жалуются на общую слабость, быструю утомляемость, пониженную работоспособность. Обычно имеются геморрагии в коже и слизистых оболочках, кровотечения из носа и десен, маточные, желудочно-кишечные и почечные кровотечения, кровоизлияния в головной мозг и любой другой орган.
Среди апластических анемий у детей наряду с приобретенными формами, существенно не отличающихся от апластических анемий у взрослых, выделяют врожденный и семейный варианты.
Анемия Фанкони, носящая семейный характер, чаще встречается среди мальчиков. Первые симптомы болезни обычно проявляются в возрасте 6—8 лет. Изменения гемопоэза сочетаются с геморрагическим синдромом и разнообразными аномалиями развития скелета и внутренних органов в виде уменьшения количества костей запястья, отсутствия или гипоплазии большого пальца, клинодактилии, микроцефалии, деформации грудной клетки, пороков развития органов дыхания, почек и мочевыводящих путей, врожденных пороков сердца и других изменений. Наблюдаются также эндокринные нарушения в виде пигментации кожи и слизистых оболочек, выраженность которой обычно соответствует тяжести течения процесса.
Врожденная парциальная апластическая анемия Джозефа — Дайемонда — Блекфена развивается в раннем детском возрасте. Возможно, она связана с аутоиммунным процессом, возникающим в период внутриутробного развития или с врожденным метаболическим дефектом. Анемия различной степени сочетается с глубокой эритро- и нормобластопенией на фоне миелокариоцитопении. Лейкопээз и тромбопоэз сохранены, не наблюдается кровоточивости.
Для семейной апластической анемии Эстрена — Дамешека характерны те же изменения со стороны кроветворения, что при апластических анемиях взрослых. В отличие от апластической анемии Фанкони при этой форме не наблюдаются аномалии развития скелета и внутренних органов.
Вопрос 25. Наследственные гемолитические анемии, классификация. Ферментопатии: классификация. Недостаточность Гл-6-ФДГ: этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
Наследственные гемолитические анемии делятся на три большие группы:
1. Мембранопатии эритроцитов с характерной морфологией клеток (сфероцитоз, эллиптоцитоз, стоматоцитоз, акантоцитоз и др.).
2. Энзимопатические (ферментопатические) анемии, или эритроцитарные энзимопатии (связанные с дефицитом ферментов пентозофосфатного цикла — глюкозо-6-фосфатдегидрогеназы и др.; связанные с дефицитом ферментов гликолиза — пируваткиназы и др.; связанные с нарушением метаболизма нуклеотидов дефицит пиримидин-5-нуклеотидазы и др.).
3. Гемоглобинопатии («качественные» гемоглобинопатии HbS, С, Д, Е и др. и «количественные» гемоглобинопатии — талассемии).
Энзимопатии обусловлены наследственным дефицитом ряда ферментов эритроцитов. В мире насчитывается несколько сотен миллионов человек (примерно 1/20 человечества) — носителей наследственного дефицита глюкозо-6-фосфатдегидрогеназы (Гл-6-ФДГ). При недостатке Гл-6-ФДГ блокируется реакция окисления глюкозо-6-фосфата в пентозофосфатном цикле, вследствие чего уменьшается образование восстановленной формы глутатиона, предохраняющего SH-группы глобина и мембраны эритроцитов от повреждающего действия различного рода окислителей. Это сопровождается снижением устойчивости эритроцитов к действию активных форм кислорода, окислительной денатурацией гемоглобина и белков мембраны эритроцитов с последующим внутрисосудистым гемолизом клеток.
Описано около 90 различных мутантных форм Гл-6-ФДГ, из которых основными являются европейская форма дефицита (активность фермента в пределах 90% от нормы), африканская (10–15%) и средиземноморская (менее 1%). Дефицит Гл-6-ФДГ наследуется как сцепленный с Х-хромосомой признак, в связи, с чем заболевают в основном мужчины.
Клинически носительство дефицита Гл-6-ФДГ проявляется острыми гемолитическими кризами при приеме некоторых лекарств, обладающих окислительными свойствами: хинин, сульфаниламиды, производные салициловой кислоты и др., при употреблении в пищу конских бобов и стручковых растений (фавизм), а также на фоне заболевания вирусным гепатитом или гриппом. В период гемолитического криза у больных выявляются признаки внутрисосудистого гемолиза — повышение температуры тела, бледность, умеренная желтушность кожи и склер, головная боль, рвота, диарея. Вследствие гемоглобинурии возможно развито острой почечной недостаточности.
Вопрос 26. Мембранопатии: классификация. Наследственный микросфероцитоз (болезнь Минковского-Шоффара): этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
Мембранопатии. Основным патогенетическим звеном гемолитических анемий этой группы является генетический дефект белково-липидной структуры мембраны эритроцитов, что приводит к изменению формы и эластичности клеток. В результате нарушается способность эритроцитов деформироваться в узких участках кровотока, в частности при переходе из межсинусных пространств селезенки в синусы. В процессе циркуляции эритроциты постепенно теряют оболочку и, в конечном счете, разрушаются макрофагами РЭС. Из группы мембранопатий наиболее часто встречаемым заболеванием является наследственный микросфероцитоз (болезнь Минковского-Шоффара), в основе которого лежит наследственный дефект белков мембраны (анкирина, спектрина, белка полосы 3, 4, 2), способствующий повышенной ее проницаемости для ионов натрия. Избыток натрия, а вместе с ним и воды увеличивает объем эритроцитов и придает им характерную шаровидную форму (сфероцитоз). Утрата части клеточной оболочки приводит к уменьшению размеров эритроцитов и образованию микросфероцитов (микросфероцитоз). В результате прогрессирующей фрагментации мембраны после двух-трех последующих прохождений через селезеночные синусы микросфероциты подвергаются внутриклеточному гемолизу. Одной из причин укорочения продолжительности жизни микросфероцитов (до 7–14 дней) служит также истощение их ферментных ресурсов (расход АТФ, глюкозы) в процессе удаления из клеток избытка воды.
Аномалия передается с аутосомной хромосомой и наследуется по доминантному типу, т.е. болезнь проявляется не только у гомозигот, но и у гетерозигот. Гемолитические кризы возникают при воздействии холода, эмоциональном стрессе, беременности, инфекциях. Центральное место в клинической картине занимают три ведущих симптома (триада Шоффара): желтуха, бледность кожи и слизистых, спленомегалия (у 75–80% больных).
Вопрос 27. Гемоглобинопатии: классификация. Талассемия: этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
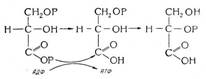
Гемоглобинопатии (гемоглобинозы) связаны с наследственным нарушением синтеза гемоглобина. «Качественные» гемоглобинопатии и сопровождаются изменением первичной структуры молекулы гемоглобина, «количественные» гемоглобинопатии характеризуются нарушением количественного соотношения НЬА и HbF в крови из-за недостаточности образования отдельных полипептидных цепей глобина. Как и носительство дефицита Гл-6-ФДГ, наследственные гемоглобинопатии относятся к числу наиболее распространенных в человеческой популяции генетических аномалий. Среди известных форм гемоглобинопатии наибольшее значение в практическом отношении представляют гемоглобиноз S (серповидно-клеточная анемия) и талассемия.
Талассемия (средиземноморская анемия) связана со снижением или отсутствием синтеза α-, β-, δ- или γ-цепей глобина. В зависимости от этого различают α -, β-, δ-, и γ-талассемию. Чаще всего встречается нарушение синтеза β-глобиновых цепей — β-талассемия. В этом случае содержание HbAl (α2β2) уменьшается, а уровень HbF (α2 γ2) и НЬА2 (α2δ2), напротив, возрастает. Недостаточный синтез β-цепей приводит к избыточному образованию α-цепей. Лишние γ-цепи способствуют появлению нестабильного гемоглобина, который преципитирует и выпадает в эритроците в виде «телец включения», придавая им форму мишеней. Кроме того, образующиеся в избытке α-цепи вступают в соединение с SH-группами мембраны и повышают ее проницаемость, нарушаются процессы ассимиляции железа и синтеза гемоглобина. Это обусловливает раннюю гибель эритроцитов в результате внутриклеточного гемолиза с развитием гипохромной анемии.
Развернутая картина тяжелой гемолитической анемии возникает при гомозиготном наследовании нарушения синтеза β-цепей — болезни Кули, проявляющейся физическим и умственным недоразвитием, бледной желтушной окраской кожи с признаками гемосидероза, придающего коже зеленовато-коричневый оттенок, деформацией костей черепа (башенный череп, увеличение верхней челюсти, нарушение прикуса; на рентгенограмме — расширение костномозгового канала трубчатых костей, поперечная исчерченность плоских костей черепа — игольчатый периостоз), язвами нижних конечностей, выраженной гепато- и спленомегалией.
Вопрос 28. Серповидноклеточная анемия:этиология, патогенез, клинические проявления, изменения в костном мозге и переферической крови.
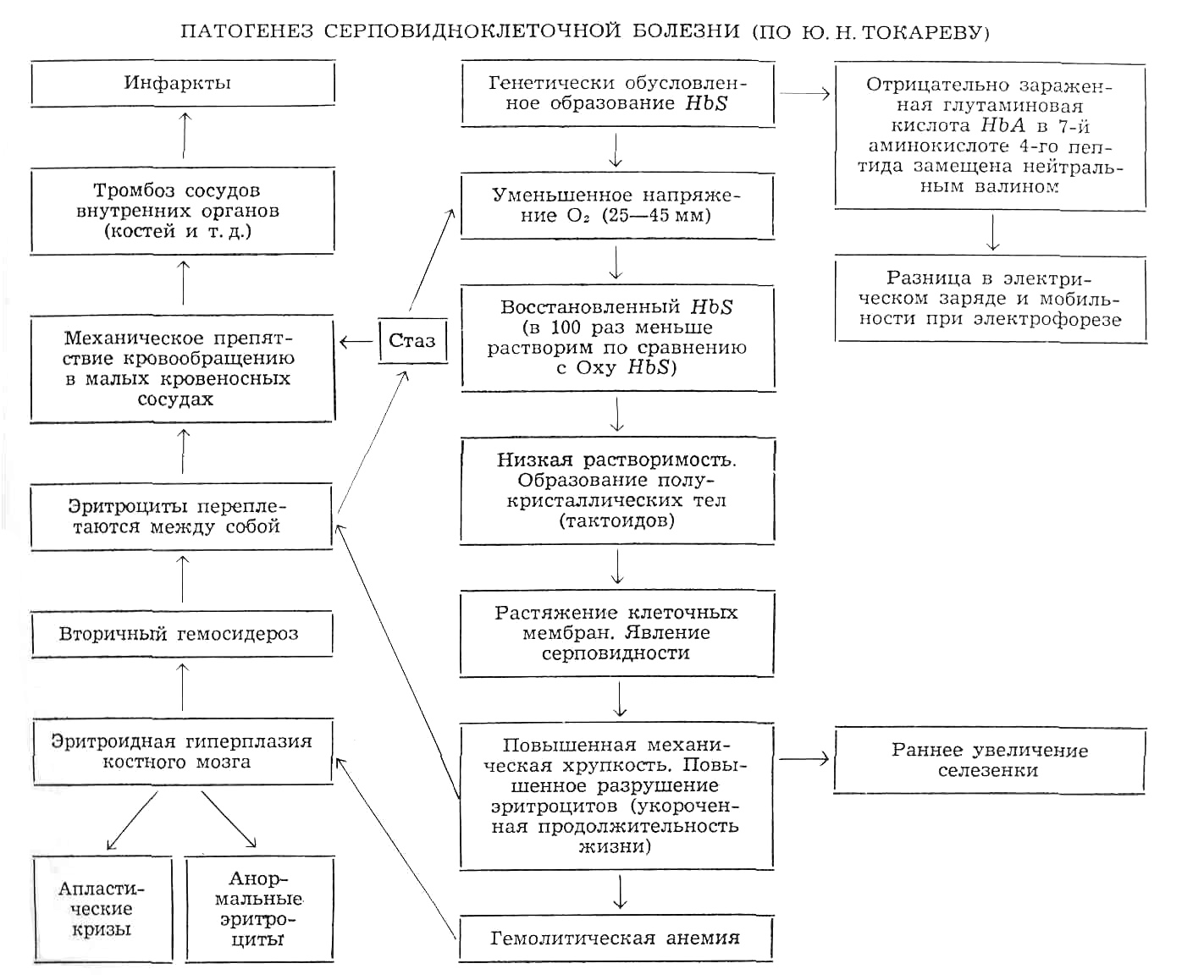
Гемоглобиноз S. Заболевание возникает в связи с наследованием патологического гемоглобина S, в котором гидрофильная глутаминовая кислота в 6-м положении β-цепи глобина замещена на гидрофобный валин. Это приводит к смене электрического заряда и полимеризации гемоглобина в условиях гипоксии, снижению его растворимости с образованием тактоидов (веретенообразных остроконечных кристаллов), которые растягивают оболочку эритроцитов. В результате клетки приобретают форму «серпа», теряют пластичность, повышают вязкость крови, замедляют кровоток, вызывают стаз. Стаз, в свою очередь, приводит к развитию гипоксемии, еще более повышая уровень «серпления» эритроцитов. Острыми концами серповидные эритроциты могут повреждать другие измененные и неизмененные эритроциты, что сопровождается внутрисосудистым гемолизом. Часть серповидных эритроцитов разрушается в селезенке. Средняя продолжительность жизни эритроцитов при серповидно-клеточной анемии не превышает 17 дней.
Тяжелая анемия проявляется лишь у гомозиготных по HbS носителей. Усиление образования серповидных эритроцитов с развитием гемолитического криза отмечается при действии низких температур, патологических состояниях, сопровождающихся ацидозом, инфекциях, дегидратации, лихорадке, голодании, заболеваниях легких, в условиях гипоксии. Вследствие компенсаторной спленомегалии у ряда пациентов в силу неизвестных причин вероятна массивная секвестрация эритроцитов в селезенке, что может стать причиной развития гипотензии и внезапного летального исхода. У гетерозигот заболевание протекает, как правило, бессимптомно. Поскольку серповидные эритроциты являются непригодными для жизнедеятельности малярийных плазмодиев, люди-носители аномального HbS — обладают резистентностью к малярии.
Патогенез серповидноклеточной болезни
Вопрос 29. Приобретенные гемолитические анемии: классификация,проявления в органах кроветворения и в периферической крови. ГБН: этиология, патогенез, характеристика клинических форм, изменения в костном мозге и периферической крови, принципы терапии и профилактики.
Приобретенные гемолитические анемии. Среди заболеваний этой группы выделяют иммунные гемолитические анемии и анемии, связанные с воздействием прямых гемолизинов и других повреждающих факторов.
Гемолитическая болезнь новорожденного (ГБН) или эритробластоз плода развивается в результате несовместимости матери и плода по антигенам эритроцитов системы Rh (D) (у Rh-положительных детей от Rh-отрицательных матерей) или по антигенам эритроцитов системы АВО (у детей с группой крови А, В или АВ, матери которых имеют группу крови 0). Первая беременность Rh-отрицательной матери Rh-положительным плодом обычно протекает нормально. Во время родов происходит иммунизация матери антигенами эритроцитов плода с выработкой антиэритроцитарных антител (анти Rh (D)-IgG), которые в период второй беременности Rh-положительным плодом фиксируются на эритроцитах плода и обусловливают гибель эритроцитарных клеток путем внутриклеточного гемолиза с развитием эритробластоза плода. Основными симптомами ГБН являются желтуха, гепато- и спленомегалия, в тяжелых случаях — отеки, асцит (из-за недостаточности кровообращения). Наиболее опасным симптомом анемии служит «ядерная желтуха» с признаками поражения нервной системы вследствие токсического действия непрямого билирубина, к которым относятся нистагм, судорожные подергивания, крик высокой тональности. Встречаются случаи мертворождения.
Трансиммунные гемолитические анемии развиваются при проникновении в организм новорожденного антиэритроцитарных антител матери, страдающей аутоиммунной гемолитической анемией.
Анемии при действии прямых гемолизинов и других повреждающих факторов -э та группа анемий объединяет гемолитические состояния, при которых полноценные в морфофункциональном отношении эритроциты разрушаются под действием различных факторов. Патогенез этих анемий различен — разрушение мембраны эритроцитов, истощение их ферментных систем и т.д.
· гемолитических (фенилгидразин, свинец, бензол, мышьяковистый водород, анилиновые красители, змеиный и грибной яды и др.),
· бактериальных (токсины гемолитического стрептококка, стафилококка и др.), паразитарных (малярия, бабезиоз).
Вопрос 30. Аутоиммунные гемолитические анемии: классификация, этиология, патогенез, спектр аутоАТ при АИГА. Характеристика пароксизмальной холодовой гемоглобинурии.